Cardiac rhabdomyomas are usually detected prenatally or during the first year of life. They account for over 60% of all primary cardiac tumors.1 Studies have demonstrated that the incidence of cardiac rhabdomyoma is 0.002-0.25% at autopsy, 0.02-0.08 % in live-born infants, and 0.12% in prenatal reviews.2 Cardiac rhabdomyomas are increasingly clinically recognized in utero because of the use of ultrasonography as part of routine prenatal screening.3 Cardiac rhabdomyomas frequently occur in association with tuberous sclerosis, an autosomal dominant inherited or sporadically occurring disorder characterized by widespread hamartomas that variably involve the brain, kidneys, heart, skin, and other organs. Fifty percent of patients with tuberous sclerosis develop a cardiac rhabdomyoma. Similarly, approximately 51-86% of children diagnosed with cardiac rhabdomyomas demonstrate clinical or radiological evidence of tuberous sclerosis or have a positive family history.4 Cardiac rhabdomyomas can occur sporadically, in association with congenital heart malformations, or in the setting of certain genetic disorders. Cardiac rhabdomyomas most frequently arise in the ventricular myocardium but may be located in the atria, epicardial surface, or cavoatrial junction. The clinical features of cardiac rhabdomyomas are extremely variable and are dependent upon the location, size, and number of tumors in the heart. Although cardiac rhabdomyomas can be associated with intrauterine fetal demise, many are detected incidentally during routine antenatal sonographic screening. Symptoms arise because of chamber or valve obstruction, arrhythmias, or failure from extensive myocardial involvement. Tumors obstructing the right-sided inflow or the outflow of the ventricles can lead to decreased cardiac output, atrial and caval hypertension, hydrops fetalis, and death. Arrhythmias, both ventricular and atrial, are not uncommon. Congestive heart failure and a low cardiac output can occur when the tumor extensively involves the myocardium or the ventricular papillary muscles. Cyanosis and decreased peripheral pulses can result. Death occurs because of obstruction of ventricular blood flow, arrhythmias, valve stenosis, or from loss of functional myocardium secondary to extensive tumor involvement. Patients with a cardiac rhabdomyoma (especially when multiple) should be examined for associated clinical and pathological features of tuberous sclerosis. The classic clinical triad of tuberous sclerosis includes mental retardation, epilepsy, and facial angiofibromas. The presence of multiple cardiac rhabdomyomas often precedes the manifestation of the cutaneous and neurological features of the disease. The physical examination of the newborn may consequently be unremarkable. However, hypopigmented "mountain ash" macules, angiofibromas, and/or retinal astrocytic tumors are early features that can occasionally be seen at birth. Other late-occurring cutaneous manifestations of tuberous sclerosis include subcutaneous nodules, subungual fibromas, linear epidermal nevi, and café au lait pigmentation. Seizures during the neonatal period are not infrequent. Imaging may reveal the presence of cortical or subependymal tubers. Renal angiomyolipomas usually manifest later in life. A detailed family history pertaining to the existence of relatives with features of tuberous sclerosis should also be obtained. Rhabdomyomas are frequently diagnosed by fetal echocardiography during the prenatal period. Multiple lesions are often associated with tuberous sclerosis. Cardiac rhabdomyomas appear as well-circumscribed homogenous hyperechoic masses that most frequently involve the ventricles but can be found at any location in the heart. Small multiple lesions may manifest as a thickened myocardium on ultrasonography and MRI. MRI is used if the results of echocardiography are inconclusive or if aggressive surgical management is being planned. On T1-weighted images, rhabdomyomas on MRI have a signal intensity similar to that of myocardium. On T2-weighted images, the signal intensity is increased. MRI is also used to evaluate the presence of tuberous sclerosis involving the brain, kidneys, and liver. The presence of cardiac rhabdomyomas (especially multiple) is strongly suspicious for tuberous sclerosis. Consequently, a detailed family history should be obtained and genetic counseling should be offered even in the absence of other signs of the disease. Echocardiography and magnetic resonance imaging are the main diagnostic tools. Cardiac catheterization is seldom required. Endomyocardial biopsy and histological assessment are the criterion standard for diagnosis. Other primary cardiac tumors of the fetus include fibroma, myxoma, teratoma, and hemangioma. On ultrasonography, rhabdomyomas appear as round homogeneous hyperechoic masses located predominantly in the ventricles and interventricular septum. They can be single or multiple. Cardiac fibromas are located in the myocardium (usually the ventricle) and often bulge into the chamber lumen. They are firm or rubbery and do not demonstrate hemorrhage or necrosis. Calcifications are typically present, a feature that distinguishes them from rhabdomyomas. The microscopic features of fibromas show dense fibroblasts and collagen. Elastic fibers can frequently be identified with the use of special stains. Fibromas are the second most common hyperechoic cardiac tumor, after rhabdomyoma. Myxomas are endocardial-based lesions that do not infiltrate into the underlying tissue. They are commonly located on the left atrial side of the interatrial septum. They can be firm and lobulated, myxoid and gelatinous, or friable and irregular. The cells of the myxoma possess an abundant eosinophilic cytoplasm and have indistinct cell borders. They arise in a myxoid background. Fibrosis, calcification, and thrombosis are common, and extramedullary hematopiesis occurs in 10% of the cases. They are only moderately echogenic on ultrasonography. Using ultrasonography to distinguish between rhabdomyoma, fibroma, and myxoma when occurring as a single cardiac lesion can be extremely challenging. However, a cardiac tumor in a young patient possessing features of tuberous sclerosis or a positive family history is almost certainly a cardiac rhabdomyoma. Distinguishing rhabdomyoma from teratomas or hemangiomas is less of a diagnostic challenge. Teratomas are extracardiac masses that are located in the pericardial cavity and are usually associated with a pericardial effusion. They are composed of both cystic and solid areas. On histology, mature derivatives of all 3 germ lines (endoderm, ectoderm, mesoderm) are found. Skeletal muscle, cartilage, liver, pancreas, and glandular tissue have all been observed in cardiac teratomas. These tissues can be readily identified on ultrasonography. Cardiac hemangiomas can occur in any chamber but are usually found in the right atrium. They can be intramural or endocardial based. The intramural hemangiomas are poorly circumscribed lesions that are hemorrhagic and congested. Endocardial hemangiomas are well-circumscribed lesions that are variably myxoid. The microscopic features of cardiac hemangiomas are diverse. They can be composed of multiple thin-walled ectatic vessels (cavernous hemangioma), multiple smaller capillarylike vessels (capillary hemangioma), or thick-walled dysplastic arteries, capillaries, and venouslike vessels (arteriovenous hemangioma). Liposarcomas may have vacuolated lipoblasts. This is an invasive tumor with features of malignancy such as tissue destruction and necrosis. Such a neoplasm would not be expected in a child. Cardiac rhabdomyomas range from 1 mm to 10 cm in greatest dimension and can occur in isolation or be multiple. They most frequently arise in the ventricular myocardium but can be located in the atria, epicardial surface, or cavoatrial junction. Larger lesions can lead to obstruction of inflow or outflow tracts. Rhabdomyomas are round or lobulated and grossly well circumscribed. Cardiac rhabdomyomas are well-circumscribed nodules that lack a capsule but are distinct from the normal surrounding myocardium. The rhabdomyoma cells are round or polygonal in shape and are enlarged with clear cytoplasm. The clear cytoplasm is secondary to loss of glycogen that occurs during standard slide preparation techniques. Glycogen reacts strongly with periodic acid Schiff (PAS) stain when adequate technique is used to prepare the slides. The myogenous origin of rhabdomyoma cells is demonstrated by the strong expression of striated muscle markers including actin, myoglobin, vimentin, and desmin. Ubiquitin, hamartin, and tuberin are also positive. Proliferation markers such as MIB-1 are negative. The most common genetic disorder associated with cardiac rhabdomyoma is tuberous sclerosis. The familial form of tuberous sclerosis is an autosomal dominant disorder characterized by widespread hamartomas variably involving the brain, kidneys, heart, skin, and other organs. Cardiac rhabdomyoma growth tends to occur up until 32 weeks gestation after which the cells lose their ability to divide and undergo apoptosis. This process is likely mediated by ubiquitin, a regulatory protein that is strongly expressed in rhabdomyoma cells. Ubiquitin stimulates the degradation of myofilaments. Apoptosis ensues and leads to the eventual regression of the hamartoma. Partial or complete resolution occurs in most cases regardless of the initial size of the tumor. The tumor is mitotically inactive and lacks the capacity to spread. No staging system exists. The natural history of cardiac rhabdomyomas is that of complete or partial regression with consequent resolution of symptoms. The reported survival rates vary from 81-92%.2 Most patients can be managed conservatively because the natural history of the rhabdomyoma is that of regression. Conservative management includes frequent monitoring with echocardiography and electrocardiograms. Patients with arrhythmias are treated with antiarrhythmic medications. If medical treatment fails to control the arrhythmias or blood flow is severely obstructed, surgical management is recommended. Regression of the tumor is associated with resolution of the arrhythmia. Surgical intervention is reserved for patients with symptoms of severe hemodynamic compromise or intractable arrhythmias. Surgery involves removal of the intracavitary portion of the tumor without complete excision of the entire lesion. FibromaCardiac Rhabdomyoma
Definition
Cardiac rhabdomyoma is the most common primary pediatric tumorof the heart and is considered to be a hamartoma of developing cardiac myocytes. These tumors may demonstrate mild atypical histologic features, but they lack the capacity for metastasis or invasion. Although the behavior of the cardiac rhabdomyoma is benign, its positioning within critical areas in the heart can lead to lethal arrhythmias and chamber obstruction. The natural history of these lesions is to spontaneously regress.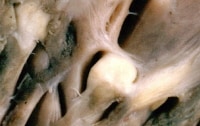
Rhabdomyoma. Grossly a white nodule in the myocardium.
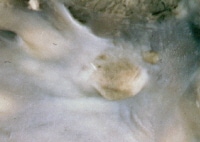
Rhabdomyoma endocardial nodule.
Epidemiology
Etiology
Sporadic mutations are responsible for more than 50% of rhabdomyomas.5 Tetralogy of Fallot, Ebstein abnormality, and hypoplastic left heart syndrome can rarely be associated with cardiac rhabdomyoma, a phenomenon that complicates both management and prognosis.
The most common genetic disorder associated with cardiac rhabdomyoma is tuberous sclerosis. Other genetic disorders associated with cardiac rhabdomyomas include Down syndrome in the setting of tuberous sclerosis and basal cell nevus syndrome.6,2,7 Location
Clinical Features
Wolff-Parkinson-White syndrome has been observed in a higher proportion of patients with tuberous sclerosis, as compared with the general population. This may be secondary to rhabdomyomas located at the atrioventricular junction. The tumor cells may act to create an accessory pathway. Regression of the tumor is associated with resolution of the arrhythmia and loss of the delta wave on electrocardiogram.
Endocardial hemangiomas are typically capillary or mixed cavernous–capillary. Intramural hemangiomas can be capillary, cavernous, or arteriovenous. Adipose or fibrous tissue can be found in intramural hemangiomas. On ultrasonography, hemangiomas demonstrate a complex echogenicity with cystic and solid areas mixed with calcifications. Gross Findings
They possess a solid tan-white homogeneous consistency and are often watery and glistening on cut surface. Calcification and hemorrhage are infrequent. Intracavitary extension can occur in as many as half of the cases.Microscopic Findings
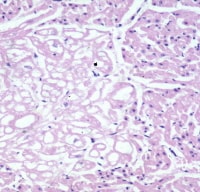
Rhabdomyoma. The tumor is seen at the left side of the image. The cells are clear compared with the normal myocardium.
The nuclei of the rhabdomyoma cells are central or eccentrically located. In some cells, eosinophilic septae stretch from the cell membrane to a centrally placed nucleus, imparting a "spiderlike" appearance to the cell. These "spider cells" are pathognomonic for cardiac rhabdomyomas and are thought to represent degenerating rhabdomyocytes (see Media file 4). They may be found scattered in small groups throughout the myocardium or clustered together forming larger nodules.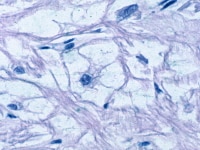
Rhabdomyoma. Microscopy showing a typical "spider" cell.
The septae of spider cells stain strongly for ubiquitin. The ubiquitin pathway facilitates myofilament degradation, intracytoplasmic glycogen vacuolization, and the production of spider cells. Spider cells then undergo apoptosis, myxoid degeneration, and regression of the rhabdomyoma. Mitotic activity is absent and fibrosis and calcification are infrequent. On electron microscopy the cytoplasm of cardiac rhabdomyoma cells are filled with nonmembrane-bound glycogen and small mitochondria. Poorly developed T tubules and fragmented sarcomeres are also present. Cellular junctions that resemble intercalated discs surround the cell periphery.Immunohistochemistry
Molecular/Genetics
Genetic analysis has identified 2 disease genes. TSC-1 on chromosome 9q34 encodes for the protein hamartin, and TSC-2 on 16p13 encodes for tuberin. These proteins are both tumor suppressor genes that appear to assist in the regulation of growth and differentiation of developing cardiomyocytes. Other genetic disorders associated with cardiac rhabdomyomas include Down syndrome in patients with tuberous sclerosis and basal cell nevus syndrome.Tumor Spread and Staging
Prognosis and Predictive Factors
The prognosis of patients with rhabdomyomas is chiefly determined by the size and location of the lesion. Tumors larger than 20 mm in diameter are more likely to cause hemodynamic disturbances and/or arrhythmias, which are associated with an increased risk of death.
Rhabdomyomas that obstruct the inflow or ventricular outflow tracts or that alter valve function and lead to regurgitation are also indicators of a poor prognosis. Differentials
Hemangioma
Lipomas
Liposarcoma
MyxomaMultimedia
Media file 1: Rhabdomyoma. Grossly a white nodule in the myocardium. Media file 2: Rhabdomyoma endocardial nodule. Media file 3: Rhabdomyoma. The tumor is seen at the left side of the image. The cells are clear compared with the normal myocardium. Media file 4: Rhabdomyoma. Microscopy showing a typical "spider" cell.
Sunday, October 17, 2010
(Enlarge Image)
(Enlarge Image)
(Enlarge Image)
Labels: Cardiac Rhabdomyoma
Subscribe to:
Post Comments (Atom)
0 comments:
Post a Comment